

Patient stories
Some of our members have generously shared their stories and experiences with porphyria over the years. It sometimes just helps to know you’re not alone.
More recently, some amazing and heartfelt stories were also shared for Porphyria Awareness Week 2020 during 8 Days of Porphyria.
Don’t forget, if you would like to make contact with someone with porphyria, you can always get in touch or come to one of our events.
Frances’ experience with AIP
My AIP occurred about the time I was a young college student studying for a diploma in Applied Biology in the early 1970s.
Feeling ill did not surprise me as I had suffered from severe asthma and severe eczema. As I was afraid to lose my place at college I soldiered on.
I had been taking Tedral tables for asthma, which contained a barbiturate to counteract an unwanted effect of ephedrine contained in the tablet. Also I had taken a few of my mother’s sleeping pills to get some sleep before my exams. These were most probably the two chief precipitating factors of my attack.
Consequently I experienced acute abdominal pain, dark red urine, (it was almost black at one point), and was extremely nervous. The nervousness was so bad I needed pin-drop silence and so was afraid of being admitted to hospital.
My hair was falling out in big bunches. My back and limbs ached. The last symptom to appear was digital extensor paralysis of my fingers except the small finger on my left hand. I was grossly underweight. It was all hell. Doctors were baffled.
Symptoms fluctuated. No one examined me when the abdominal pain was at its worst and I was thrashing about in the bed. I was variously believed to have hysteria, anorexia, appendicitis and even Hodgkin’s disease. My appendix was removed and biopsies taken. The results of an EMG indicated a neurological abnormality.
Then my father advised my urine be tested for AIP, as his brother had been extremely ill with this. The first two times the result was negative. Mother pleaded with hospital staff to do the test again. The result of the third test was positive. Mother and I shared a private joke. We would say I had gone ‘APE’ again when I was ill -’A’ standing for asthma, the ‘P’ for porphyria and the ‘E’ for eczema.
It took me several years to recover from my attack. Essentially, it cleared on its own, once I knew what substances to avoid and started eating more. Eventually I worked as a medical technician for a few years.
My main health problem since my attack has been severe eczema for which I have had about nine hundred injections ACTH (a powerful hormone) spread over twelve years. These days I have a son at university. My porphyria does not interfere much with my life.
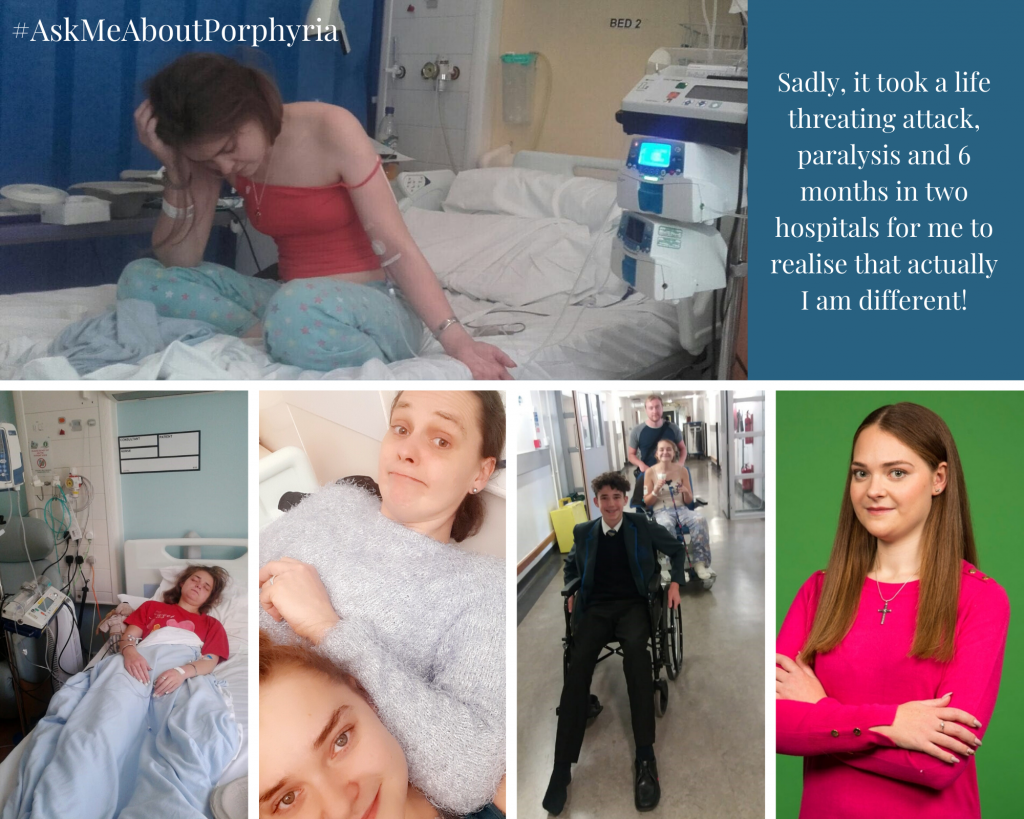
Elizabeth’s story
Throughout my childhood, I thought I was going to travel the world, but when I developed symptoms of acute intermittent porphyria at 18, my dreams were dampened slightly.
Fierce pain, paralysis and sickness took over my body, and for week’s doctors seemed to think I was either making it up, mad or anorexic. Eventually they discovered it was porphyria, and it was inherited from my father. We had never heard of it, as my dad had never suffered symptoms, and neither had most of the medical staff. We began a very steep learning curve, as did the doctors.
We learned that antibiotics prescribed for a minor infection, had probably triggered it in my case, but after that, I generally suffered attacks in my pre-menstrual period. These attacks varied in severity, but the pain and vomiting were omnipresent features. My weight gradually dropped, as I was unable to regain it in the short periods between attacks and eventually these monthly attacks took their toll. I had a severe attack that led to almost complete paralysis, which meant I needed assistance to breathe and a feeding tube. This time was frightening for me and terrible for my family, but the nursing staff in Intensive Care, were very reassuring. Over the next 18 months, the paralysis subsided and I returned to my regular monthly attacks.
Although able to bring me out of attacks with haem arginate, the doctors struggled to try and stop the attacks happening at all. After many experimental treatments, we have in the last couple of years hit on one that appears to be working. I now have haem arginate on a weekly basis and I have only had a few attacks this year.
Although still a bit wobbly when walking, I am only 29, and after 11 years of ups and downs, I have just got a Masters degree and I am now trying to set up a small freelance proofreading business. I want to emphasise that without my family to encourage me and care for me, I would never have got to this point of looking to the future with optimism.
Recent update: now 37, Elizabeth has not had any severe attacks that require hospitalisation for many years. She has a three-year old child and has been successful with her editing business, which goes from strength to strength each year. Currently still receiving haem arginate on a weekly basis, Elizabeth has also achieved her ambition of travelling to a wide variety of countries, albeit developed ones that could deal with such a complicated condition if necessary.
Alan and AIP
In 1969, when I was 19, I had terrible stomach pains at work. My doctor prescribed laxatives, which were useless. After a week of pains my mother persuaded him to admit me into hospital, as she had had a burst appendix as a child. In the hospital I was left for a week with the pains until my appendix was finally taken out. But to no avail. The pains continued.
At this point I’d been given anaesthetics, antibiotics and even sleeping tablets every night. Slowly I became weaker, until I was unable to move any of my limbs. During this time I was not believed by any of the members of staff, even when I was unable to get up off the floor when I had fallen out of bed. I even convinced myself that there was something wrong in my mind after they had a psychiatrist come and evaluate me.
Suddenly after four weeks my consultant arrived at my bedside and informed me that he wanted to do a lumber puncture on me, which involved inserting a long needle up my spine to extract fluid.
Within two hours I was told that I had Porphyria. I remember a lot of fuss was made to get me ready to be shipped off to Manchester Royal Infirmary, where I stayed for the next five months. Here I was believed at last, although the damage was already done, regarding all the drugs that I shouldn’t have had.
As it was early days in the treatment of Porphyria I was willingly used as a guinea pig for various tests to try and understand the condition. Eventually I got stronger, learned how to walk again, regained a lot of muscle tissue, having gone from eleven down to seven stones, and came home.
Looking back all those years it was the worst episode of my life. I don’t blame the first hospital for not knowing about porphyria, as we know now it’s quite a rare condition. It transpired months later that my first consultant just happened to mention my symptoms to a colleague in the hospital canteen over coffee. Thank goodness, this colleague, whom I have always owed my life to, knew of the symptoms of porphyria.
Linda’s story: AIP
In my late teens I was often unwell, violent stomach pains constant vomiting followed by excessive tiredness, mood change and depression. Is it drugs or is she anorexic they asked? No it’s her life style; she’ll grow out of it they said. How wrong and how right they were, drugs? No, anorexia? No, but crazy teen lifestyle that could have been potentially life threatening? Yes.
Years passed I married and became pregnant, joy turned to fear as Ectopic pregnancy, then Appendicitis was diagnosed, miscarriage seemed inevitable if surgery took place. A glucose drip was provided, to aid re-hydration in preparation for surgery. Within hours a decline of symptoms was observed, within days a full recovery, the pregnancy continued and a healthy baby boy was born.
Two years later following the birth of my second son, I was immediately admitted back into hospital with suspected food poisoning. Following administration of an intravenous drip, to aid re hydration a recovery quickly occurred. Diagnosis was never clarified. With two young children, foster parent to two more, a husband away a lot life was pretty hectic. I began to suspect I may be anaemic or have postnatal depression, I had similar symptoms.
My GP although supportive in the beginning, after tests proved inconclusive basically told me to pull myself together, I was a healthy young woman, there was nothing wrong with me.
I began to feel very low, began to think I was going mad, that I must be a hypochondriac, was I imagining the feelings that I was experiencing? The weakness in my limbs, the shakes the palpitations and breathless, the stomach and back pain. What was making me vomit? Why did I feel the sun was shining one minute and the sky was grey the next.
The answer arrived, my cousin who had recently given birth to her third child following a considerable gap from her other two children, had became unexplainably critically ill, relatives were informed, an aunt who had been abroad came to visit her niece.
My aunt had years earlier been diagnosed as having acute intermittent Porphyria, she had been advised not to have children and had moved abroad with her husband who was in the forces. The effects and treatment of her illness were never discussed.
On seeing her niece my aunt immediately suggested Porphyria as the cause, the medical team sought advice; suitable treatment was given and after a considerable length of time my cousin made a full recovery.
Following this diagnosis and a progress in medical science the entire family (which is considerably large) were invited for screening. Consequently tests, not DNA which was unavailable at the time, confirmed that I, like many others in my family have AIP. It was a relief that a name had been given as it probably accounted for many of the symptoms that I was experiencing. I did not then realise how little was known or how hard it would be to obtain advice or information on managing AIP. Although I had many relatives with the same condition, many were latent sufferers and some in denial, until recently mental health implications made the illness a taboo subject in the family.
Following diagnosis I began to suffer considerable stomach and back pain gynaecological complications were diagnosed, I had a hysterectomy at the age of 28. Surgery was performed without consideration being given to my inherited condition, I was consequently very ill. (It has subsequently been questioned as to the need of such radical surgery.) Time passed although often unwell my own and medical awareness began to grow. Information via the Internet and appointments with specialists helped me to begin to understand and make informed decisions with regards to management of my condition. Joining the BPA original committee group eased isolation.
During my late thirties I was extremely unwell, often needing to be admitted into hospital. I was working in education and had been for the past 10 years. Schools have a rigid timetable, responsibility, makes you push on. Something had to give. I ended up changing my job, which was not easy. I am now employed as an ‘outreach project worker’ for children and families, an equally demanding job. However, I have learnt to prioritise and this type of work allows me to do just that. I can alter appointments and schedules to fit in with any symptoms I may be experiencing. I have learnt to listen to my body and I have also become better at saying ‘no’ and ‘slowing down’, whereas before I only stopped when ill.
I am now in my mid 40s taking HRT and possibly completing the menopause things are looking good. Yes I often feel ‘ a bit unwell’, but am no longer a regular in-patient at our local hospital. Biological or environmental, who knows? Whatever the reason I recommend getting older, it certainly appears to agree with me and lots of other AIP sufferers that I have spoken to. Some older members of the family, in their mature years, can even tolerate the odd glass of alcohol without effect, so to all AIP sufferers I would just like to say ‘don’t look back, just look forward to getting older’. CHEERS
Kerry’s experience
My mum became very poorly when she was a teenager, so poorly doctors thought she was going to die. Eventually she was diagnosed through tests as having AIP. I as a teenager, myself having suffered many bouts of severe stomach cramps, sickness and muscle pains (sometimes unable to even move my eyes) was also diagnosed as having AIP after being tested on and off for years. It took my brother to be injured in a car accident and a new doctor from a different department (unaware of the possibility of him having AIP) seeing something unusual in his blood to diagnose him also with AIP. A doctor who was seeing my family decided a few years ago that the results were wrong as we had not returned any positive results for a while. This I found shocking – as you yourself will know, borderline and negative results can be achieved in people with AIP. My mum, having had to go through a hysterectomy and being advised to discontinue her pregnancy with my brother, found this unbelievable. Myself and my mother went to Glasgow for a second opinion, where the doctor concluded that he could not understand why our Aberdeen doctor, after all these years and us having all tested positive at different times and all having had symptoms of AIP, would be happy for us to take medication that was not acceptable with AIP. Ourselves, with the help of our main doctors practice, ensure all medications are safe with AIP. My main concern now is for my 3 children and my 2 nieces, as no-one has made any contact about testing them since the genetics doctor at Aberdeen. I mentioned when my daughter was born that I had tested positive for AIP and they contacted the genetics department. They said they will wait and test her when she is older . My sons are 14 and 12 and my daughter now 3. I myself as well as my mother and brother had blood, urine and stool sampling. We ourselves used to be contacted to be tested regularly, especially myself and my mother but all communication has stopped since we asked for a second opinion. We were told we no longer needed to attend and have been unable to discuss what the opinion of the doctor in Glasgow was. We do continue to have symptoms regularly especially during menstrual time. I receive painkillers from my doctor that just about take the edge off the pain and avoid anything that could cause an attack as much as I can.
Beth’s story: Living with EPP
My name is Beth and I have suffered with EPP for my whole life. However it wasn’t until I came to my first BPA meeting in Leeds in June 2008 (aged 34) that I met others who also suffer from EPP. Like many who suffer from any type of Porphyria I have always felt quite isolated and felt that no-one (even close family) really knew what this meant.
The reason why I am writing this article is that I want you all to feel that you are not alone and also that it is not all doom and gloom having Porphyria. I have already met some nice people through the BPA!
I was born and bred in Yorkshire and brought up with two older brothers. It wasn’t until I was about 3-4 years old after lots of nagging at doctors that my parents were eventually put in touch with a consultant who diagnosed me with EPP.
I guess like many young people with EPP I felt conscious of wearing long sleeve clothes or having to put up an umbrella or even stay inside. What I can say is that suncreams and UV clothing have come a long way since I was a child. I remember being about 10 years old and having to wear this bright pink zinc cream (like cricketers wear) on my face and hands. It looked like I had pink cement put on my face!!
However my family didn’t wrap me up in cotton wool and keep me indoors all the time. Instead we went on holidays to Europe most years. Although this sounds harsh it taught me how to enjoy travelling overseas but also how to avoid the sun at the same time. Therefore I swam in the sea at 5pm at night when everyone else was leaving the beach and made more of the evenings.
This gave me a keen interest in travel and for a few years I worked for the British Tourist Authority in London promoting Britain overseas. This meant I had to travel to the USA and Europe for work. I just had to work things out my way for doing exhibitions outside. They were always in the shady areas! I have continued to work on outdoor events such as Food & Drink Festivals and the likes in my present role as Halifax Town Centre Manager. Fellow EPP sufferers must think I am mad!!
In order to help me with EPP and the outdoor events I do I have decided this year to undertake the desensitivity treatment to allow me to hopefully stay in the sun longer this Summer. I talked to some fellow sufferers at the BPA meeting in London in October last year and they recommended the treatment. However I said to Dr Sarkany that I would be unable to get to London or Dundee (where I knew they offered the treatment) for several weeks in the Spring due to work commitments. Dr Sarkany mentioned that I could be treated at my local hospital and if they didn’t know how to undertake the treatment he would send them information. As promised he did.
As I am writing this, I have just had my 5th session of treatment on what I call a vertical sunbed and at each session the time and intensity has increased. I am now up to approx 9 seconds. My family laugh when I say I’ve been to hospital for 9 seconds of treatment as it takes me longer to get changed! – it was 4 seconds initially!! I will continue to go for the next 7 weeks and hopefully this will help me in the summer.
I wanted to help other Porphyria sufferers and hence joined the BPA Committee to help with marketing and PR. My first thing I have done is set up a Facebook site group called British Porphyria Association. The reason for this is that I think Porphyria sufferers need a channel to liaise with each other whenever we so desire or need to pick each other’s brains about something or even to give us a “pick me up” when we are feeling a little down. You don’t even have to be based in Britain! Join us on Facebook.
Showing EPP who’s boss: Beth V
I was not diagnosed with EPP until I was 12. Looking back, I did have symptoms that I can remember from aged 6. My feet used to hurt and my hands used to tingle when I was out in the sun, but I didn’t think anything of it. I didn’t really go out in the sun much as I have red hair and blue eyes. My first big reaction was when my face became swollen aged 7. Mam took me to A&E, but they thought it was an allergic reaction to something I had eaten.
We went on holiday to Spain when I was 11, it was August and boiling hot. My hands began to tingle and then the pain started. It is hard to explain, but feels a bit like your skin is on fire. There was nothing to see on my hands at first, but after a couple of days the swelling started. I was in agony and kept trying to cool my hands down with cold water and ice. I couldn’t bear anyone to touch them as the pain was so bad. Mam tried to get medical advice, but they gave me steroid cream and said that I was allergic to the sun cream. When Mam rubbed it on my hands I screamed as the pain was terrible. We stayed in for the rest of the holiday as I couldn’t bear to be outside. I couldn’t wait to get home.
When I came home my hands stopped hurting so much after a couple of days, but the skin was dry and cracked, this took weeks to get better. I was told that it was an allergic reaction, and I hoped it would never happen again.
The year after, I was in the garden in May when the tingling started again on my hands; Mam took me to the doctors and although he had no idea what was wrong, he referred me to a dermatologist. I was very lucky, as the dermatologist had seen one case of EPP before and was 99% certain that this is what I had. She told Mam to cancel our holiday to Florida and sent me for blood tests. Six weeks later I was diagnosed with EPP.
I loved being outside, and my hobbies were horse riding, running and being out in the country with my dogs. I was so sad because I was told that I would not be able to go out in the sun, but I loved being outdoors.
School trips were difficult, I went to an outward bound centre with school in July; it was very hot and all the activities that they did I loved doing. I started to get ill after a day and was told to sit in the dormitory. I had nothing to do during the day. The school would not let me phone Mam to go home. I also went camping with Guides and became ill again, this time I was told to sit in a hot tent and go to sleep. I was in pain and couldn’t sleep anyway, eventually I persuaded them to call Mam and she came out of work to collect me.
I thought that this was going to be how it would be for the rest of my life. But I have been very fortunate to meet someone with EPP who lives close by who has inspired me to show EPP who’s boss. He is called John Clark and he still goes outside in the sun, and runs ultra marathons, walks in the lakes, skis and lots of other mad things.
I went on my first skiing holiday this year, John helped me with my clothing to make sure I would not be ill, as the sun reflects off the snow and can make people with my condition very poorly. I learned to ski, had a fabulous time and because of John’s help I did not get any reaction. The best thing about skiing is that I can look exactly the same as everyone else and no one would ever know that I have EPP. If I go to a sunny country I have to wear big hats, long sleeves and trousers and could never wear sandals. I feel like people are looking at me, but with skiing we all look the same. I want to go back every year.
I have also discovered climbing, although it is indoors I like to think that I would be able to do this outside. This year I am going to run my first race, with the help of John, who has said he would run with me. I still go horse riding, as I can wear my hat, riding gloves and jodhpurs so I am covered up and look like every other horse rider.
When I first got diagnosed with EPP I felt very sad and thought that my life would change forever. My life has changed forever, but it is better now. I would not like people to feel sorry for me, as EPP has introduced me to John and he has taught me that EPP is part of me and I should never feel sad about that.
Sarah’s story: EPP
When I was born back in May 1970 I was a miserable baby, being a summer baby my parents used to put me in my pram in the sunny garden little knowing that it was probably that making me miserable! No one knew what was wrong with me and why do I keep waving my arms around?! I was diagnosed as having hay-fever, which my parents thought wasn’t the case but thought the doctors knew more!
When I was four I was finally given the diagnosis of having Erythropoietic Protoporphyria – a condition which even to this day I can’t say, let alone spell..!
Well, what does having EPP mean to me, in a nutshell it means not being exposed to the sun. Luckily for me I live in England and we don’t have all that much sun here! I am not able to go in any bright light (UV) which makes the summer the worse time of year although really anytime from March until November is difficult. I use factor 60 sun screen, an umbrella and cover up with long sleeves, wear trousers etc – which all makes me very hot and irritable, I can tell you!
When I tell people I can’t go in the sun the first thing they ask is “where do you go on holiday?” I always reply, in the winter in England! But I don’t really miss holidays it’s simple things I miss or get annoyed at not being able to do, like being able to take my kids on summer picnics (during the day as opposed to going in the late evening or in winter!), being able to watch their sports days at school and even chatting with other mums at the school gate. Late night shopping is great during the summer for me as it means I can load and unload the car slowly rather than throwing things in the boot and rushing indoors! I suppose more people are aware that the sun is bad for everyone so it means there are items such as high factor sun creams available in high street chemists now. We also have the big shopping malls all indoors and undercover so I can still shop…!!
When I have caught the sun I’m in a lot of pain and discomfort, my skin feels like its burning but at the same time I don’t like it getting cold or too hot, anywhere I have been exposed swells up – it also zaps my energy and I feel so tired, I tend to lay down with an electric fan blowing cool air on me. My hands have toughened up over the years and they manage to cope with a bit of sun. A couple of years ago I caught the sun pretty bad in October in England and my face scabbed, leaving a few scars.
I have two children, Ryan (13) and Eloise (5) and they don’t have this condition, which is great and the only time it affects them is the embarrassment of walking with me and my brolly! I suppose over the years I have learnt to cope with this and being a person ‘whose glass is _ full’ I always think that there are so many other ailments and problems people have that not being able to go in the sunlight really isn’t a big deal.
The future for me will be bright, I will be spending it outdoors, riding, running, climbing and walking with my dogs, whether the sun is shining or not. I am determined to run my first ultra marathon at 18, and I will do it. If I do get ill I will just go home and think of all the fun that I have had that day and not regret one minute of it. We all get ill sometimes, so how am I any different to anyone else? There are many people in the world who have conditions much worse than me. I sometimes get a bit down and sad, but Mam and my little sister, Cait, won’t let me be sad for long.
Mam and John have made a big difference to my life. I don’t tell many people about my EPP, as they don’t understand. The people important to me do understand and that’s all that matters.
Sean’s story: EPP
Even today, my memory goes back as far as my first experience with porphyria, as it was a painful one. I was in Spain, on a family holiday. My parents noticed me fidgeting and hiding in the shade. My legs, face and hands were on fire. The only relief from pain was a temporary one when I was lying in a cold bath. As soon as I came out of the cold water, the pain came back.
I was up and down all night, in and out of the bath. The next day, the swelling of the skin began. My hands, face and legs doubled in size.
This was continuous throughout my life – every summer on a couple of occasions, my skin would burst open.
The worst attack was last year when I became 40. I was at the races and it was red hot. I was exposed for about 2 hours. The pain was so intense I had to grind my teeth – it was agony. When I woke up the next day, I resembled elephant man. My eyes were shut and my forehead was really swollen. Under my fingernails were boiling.
Throughout my teenage years were the worst, as when my friends would knock for me, I could never come out. Nobody understood. Although I’ve come to terms with it, nothing seems to help, just one simple rule. Don’t go out in the sun.
Hazel’s story
My name is Hazel. I had my 70th Birthday this year. I am an EPP sufferer.
I attended the Peterborough BPA Conference this October – the first I have ever attended.
I’ve always wanted to write my story, to tell of my experience of living with EPP, if only to help some parent with a child who is suffering from the same complaint.
I never thought anyone would be interested enough to listen until now. Hearing the very emotional video at the conference of an EPP sufferer, and the heartfelt experience of the husband of an AIP sufferer and the pain they both suffer directly, or because of a loved one, I now feel it is the right time to speak of my own experiences.
Back in the 50s when I was a child I don’t think EPP was even a known condition. It was not until 1975 at the age of 26/27 years that I was diagnosed by Professor Rowell at the Leeds General Infirmary and even back then it was a fight to get to see a specialist.
I have to start at the beginning, or at least as far back as I can remember. From the age of six or seven years I knew I was different. I don’t remember much before then about the pain so maybe that’s when it started. All I remember was dreading the summer months.
Spring was my favourite time of the year, when the earth woke up, pushing the daffodils and tulips out of the cold ground. The trees just started to pop with green shoots reaching out of the bare branches. Longer days with respite from the cold, another icy winter of the 50s was over, and I felt excited at the few days of mild warmth I could experience before my freedom would once again be curtailed.
I had just moved with my Mum, Dad, older sister and younger brother to Ilkley. We moved there with my father’s work – he was a West Riding Police Officer who was moved around every few years.
I loved living in Ilkley, it was the most wonderful place. The river Wharfe running almost at the bottom of our street, and the Cow and Calf rocks up on the moors just a mile away. Lots of green lush places around us. It was a child’s paradise.
I had not been at the Primary School very long before I was assessed, my parents were told I needed to be taught in the prefabricated buildings by Miss Paley (who was classed as a remedial teacher for under achievers), as I was, in their opinion, “not bright enough to be taught in the mainstream classes”. Even at seven years old this was quite a blow, especially as my sister was clever enough to attend the local Ilkley Grammar school. I felt like a dunce.
Every day I would walk to school and back four times, as I went home for lunch. School was probably three quarters of a mile each way, this was fine through the Autumn and Winter and wonderful at the start of Spring, but then the inevitable sun would appear and with the sun something bad started to happen. When I look back, I wonder how no one connected the dots. I knew even from a very early age it was the sun that caused my pain…why could no one else see it.
My mother took me to the doctors several times throughout the summer. I don’t remember going but I do remember my mother telling my dad that the doctors said it was all in my imagination. Another time when my hands had broken open in sores she thought it was due to a cat scratch, only I knew the truth as I prayed for rain.
How I ever learned anything at school is a mystery to me. Throughout the long hot summers of the 50s, I spent my time with my hands inside my cardigan or behind my back on the way to school to avoid the sun, which never worked. From April until October, when the sun came out I suffered.
When I arrived at school I would put the backs of my hands on the cold metal under the school desks to try for some relief. When that did not work, I would perpetually put my hand up with a ‘Please Miss may I use the toilet’…. Miss Paley must have thought I had a bladder problem! I would rush to the sink and run my burning hands under the cold tap which made them itch and tingle, so I would flap them about like a ducks wings or even press them with my other hand to get relief… I could not bear anyone to touch my hands, something as simple as a scratch felt like knives were ripping my skin. I never told my school friends I thought they would think I was making it up. Even though at times the backs of my hands would break open and bleed.
Playtimes were spent in the shadow of the prefabricated building, and at the end of the day I would run home and spend the rest of the day and night wetting flannels and putting them over my hands as a way of easing the pain, just so I could get a few hours respite and some restless sleep. I was exhausted, and the hot days seemed never ending like the pain. I would pray for rain.
I had many friends at Ilkley and I just wanted to be like everyone else. I wanted to play out, to go down to the river, cross the stepping stones and run in the fields. I would always seek the shade of the trees, I loved the trees, but it was not enough, and if I wanted to play out I had to suffer, and so it went on year after year after year.
We moved again to Kirkburton, near Huddersfield, just before my 11 plus. Needless to say, I failed that! I had by that time realized that I was not an absolute dunce as I quite enjoyed English and Maths and seemed to be quite good at it. Nonetheless I was assigned to class 1B. I am relieved to say that despite my problems I attained top of the class when I reached 2B and was sent up to 2A. I spent the rest of my school life in the A stream, even though I was near the bottom I never went back down.
I’m not saying that was the end of it. It would take a further 15 painful and depressing years before I had my diagnosis. But that is another story.
Lianne’s story: HCP
I’m 37 and was given my diagnosis of HCP at the age of 21. The first acute attack that left me hospitalised was in April 1996; I was admitted with acute abdominal pains, but after investigations found no apparent cause so I was sent home. The abdominal pains continued and I began having prolonged convulsions throughout each day. Again, being admitted to hospital (a week after being discharged) nothing was diagnosed and I was discharged again. The abdominal pains worsened, accompanied by nausea and constipation and convulsions lasting for up to 5 minutes at a time. At this point I had severe muscle weakness and had increasing difficulty walking, not only that, but my speech began to slur and stutter. My speech and mobility continued to worsen, so my parents at this stage brought me home from university and took me to my GP. After checking in my eyes and doing other examinations I was admitted to hospital with a suspected brain tumour. CT scans revealed this was not the case, but I was now completely paralysed and could only stutter a few words. I had every test and scan imaginable, but was told it all must be stress related to me being a student and holding down several part-time jobs. They stopped the medication I was taking (mefenamic acid for bad period pains) and over the coming weeks my speech returned and with physio learnt to walk again.
I returned to university, although my health had its ups and down, mainly my limbs being jerky and speech problems. If the abdominal pains got bad and I had convulsions I would suffer at home as I’d been told it was stress. Nearly 7 months later I received a phone call from the hospital asking me to come in as some mislaid blood tests had been found from when I was admitted in April. When I arrived at the hospital, in not so good health, I was admitted and spent weeks having neurological tests done. I was asked if anyone in my family had Porphyria. I’d never heard of it. My urine was sent to Cardiff for analysis to determine what type I had. It was a few weeks until I was told it was Hereditary Coproporphyria, but I remember the relief at being told what it was. I finally realised I wasn’t going mad and everything began to make sense. I was also told that mefenamic acid was the trigger.
Due to the nerve damage from so many attacks it took until 2003 before my nerve conduction tests showed a complete recovery. However the nerves in my arms and hands were very poor, so in 2004 had carpal tunnel surgery on both hands. My porphyria settled down for a few years and only a handful of hospital admissions took place between 2004-2011, for which I was treated with glucose drips and sent home.
A lot of the time in recent years I would have attacks (in line with my menstrual cycle), but having a young family I would not go into hospital. I’d meet with Dr Badminton a few times a year and he advised my GP to look into other possible health problems that could be the trigger for my attacks, as I have always stuck to my safe list of drugs and have avoided alcohol since being diagnosed. In January 2011, I was referred to a gynaecologist as my period had not stopped for 3 months and was found to have a fibroid (1.5cm) and this could be the cause of the attacks. It was recommended I have hormone treatment to suppress my ovaries, but due to my GP not informing me of this I continued to have monthly attacks, by January 2012 they had got so bad that I agreed to stay in hospital. Tests revealed my fibroid was now 10cm (and growing rapidly) and I also had endometriosis; they began Prostap injections to try to control my hormones and I was told I would need a total hysterectomy including removing my ovaries.
It is now June 2012 and I have had an acute attack every month this year. In February my haematologist got involved and for the first time I had haem arginate treatment. I have had a course of 4 treatments each month via a central line and it really makes a difference. In April I had surgery to remove my womb and ovaries, but am still having acute attacks. I have nerve damage again from so many acute attacks and will soon get test results to reveal the extent of the damage. There has been mention of me having a portacath inserted to allow me to have weekly haem arginate, so that it is a preventative measure, rather than waiting for the attack to start before administering the haem arginate. I feel very lucky to have such a good team of haematologists who have taken a keen interest in Porphyria and liaise well with Dr Badminton to ensure I get the correct support and treatment, and I’m not afraid to go into hospital at the first sign of an attack. I have a six-year-old daughter who has inherited HCP, but I hope and pray that she never has an acute attack as my twin sister, elder brother and mother (all with HCP) have been fortunate never to have suffered.
Knowledge is key, and more often than not we, as patients, are the first point of reference for information about Porphyria. I always remember to carry a safe list of drugs where ever I go!
Paula’s PCT story
After suffering with skin traumas and blistering to my hands, I was diagnosed with PCT in 2015. Unfortunately it took well over 12 months to then see a consultant dermatologist who initially refused treatment. My ferritin levels were at 398 mg and during the next 6 months I worked to reduce those levels through diet and exercise. At my next dermatology visit I was referred for venesection after successfully lowering my ferritin levels to 175 mg. Following two venesections my levels were reduced further to a “normal” 38 mg. I have seen a great improvement in my skin sensitivity to overhead fluorescent lights in the office and a slight improvement in the skin on my hands. I am assured by the dermatologist that over time my skin will improve and thicken back to it’s previous levels.
Sandra’s PCT story
My name is Sandra and I am 51 years old. I was diagnosed with Porphyria Cutanea Tarda (PCT) in September 2003. I was 45 years old and fit as a fiddle when I noticed that my urine was purple; I’ve had a hysterectomy so I was baffled and went to my GP. He tested my urine and said that I had blood, sugar and protein in my urine. He gave me antibiotics for an infection, although I had no pain anywhere. I completed the course but the symptom continued.
One day my urine was deep red and I panicked. I bottled some and took it to the doctor. Again he tested it and gave me more antibiotics. To be honest, although I knew it wasn’t right, I let it go for a few weeks. I also had facial hair growing, which I put down to taking HRT, but I was going about my normal daily life and getting excited about my forthcoming holiday to Tenerife. Around 3 days before my holiday I noticed a blister on my hand. I knew that I hadn’t hurt myself but I wasn’t worried as it was only a blister.
I boarded the plane and to be honest the blister was annoying and was getting bigger, so I decided to release the fluid with a sterilised needle in the hope that it would heal. While on holiday the blister would not heal and was throbbing; I put it down to the heat and thought it would heal when I got home to Scotland.
Within a couple of weeks of returning both hands were throbbing painfully and clusters of blisters were grouping all over my hands. I returned to my doctor who gave me creams. It got to the stage that when I was driving, the heat coming through the windscreen caused terrible pain in my hands, which were now a mess and my skin had become very fragile. The least knock and the skin ripped. Then marks started to appear on my face and feet; my feet were so swollen I couldn’t wear shoes. I was a mess and in pain.
I was tired and didn’t feel or look like me. I was feeling old. I was back and forth to my GP, who didn’t have any answers either. We were both baffled until one day I said, “Look, don’t think I’m daft, but could there be a connection between my urine and this blistering?” Don’t ask me what made me think of this but these were the only things occurring that I’d never had before. He said, “Sandra will you leave this with me? I’ll take your files home tonight and have a look.”
At 9am the next morning he phoned: “Sandra I think you have a metabolic disorder, I’ve made you an appointment at the hospital for tomorrow morning.” To cut this story short, consultants and doctors came and went looking at my hands checking my urine. “No need for a biopsy”, one doctor said, “it’s Porphyria Cutanea Tarda, she has milia”, pointing at my hands. I was bewildered. What were they talking about? One consultant briefly explained it to me; all I remember him saying was something about porphyrins, ferritin levels and start venesection straight away. This is where they remove 500mls of blood from you and repeat it periodically until the ferritin level decreases.
I was told to stay out of the light and avoid alcohol and any medication. I felt that my life had been dragged from under me.
It’s important for me to say that within one year of diagnosis of PCT I was diagnosed with Haemochromatosis, a blood condition that I was born with but had no indication of. This condition produces and stores iron in the blood and it attacks the main organs if not detected; my liver was partly damaged, but has repaired itself through venesection as my ferritin level reduced.
Haemochromatosis is the cause for my PCT which is the only porphyria which is acquired. Family should be tested for the faulty gene which causes Haemochromatosis as this can be passed on through generations.
I was so alone and frightened and had many questions, but then I heard about the BPA. The BPA showed compassion, understanding and empathy, they listened to me and gave me answers to my questions. A couple of years down the line I wondered whether there were other people feeling the way I did. With help from the BPA I managed to attend an event held in Leeds. It was amazing to see other people with porphyria. I decided to join the committee in order to start up a support group in Scotland. Special thanks to all of the committee for the work they tirelessly volunteer to do, giving up their own free time.
PS – If something is worrying you, then it’s not silly. Go to your GP.
Lynn: VP and hormone suppression
Lynn was diagnosed in her late teens with Variegate Porphyria. She had a very bad attack and, though it took some time, this led to the diagnosis. Her family hoped that she would have no further attacks, but this was not to be. In the ten years since, she has had attacks about every two years, usually in the summer months. Her family also noticed that the attacks started around the end of her menstrual cycle, and that she was regularly unwell at the same time of the month.
Her consultant decided that this was worth investigating and she had to collect urine samples every other day for over a month, to be tested for porphyrins. The samples had to be wrapped in foil to keep them dark, and then frozen to prevent them deteriorating. A special box had to be kept in the freezer, so she could store them to deliver to the lab in weekly batches. Fortunately no-one mistook them for ice lollies!
The results showed a definite fluctuation in porphyrin levels over her cycle – a fairly common pattern in female porphyrics. So the consultant referred her to a gynaecologist, to discuss hormone suppression (GnRH inhibitors). She decided to go ahead with this and the gynaecologist prescribed the first injection, which lasts a month, and wrote to her GP about follow-up injections. They have to be repeated very promptly, or the normal cycle reasserts itself.
Unfortunately, the injection caused the worst type of menopausal symptoms. She got such severe night sweats that she had difficulty getting enough sleep, something which did not surprise her mother, after her own experience of the menopause. Lynn’s GP prescribed oestrogen patches, which helped a bit, but she was glad to go back to the gynaecology clinic for advice on higher-dose, constant level HRT pills.
These helped a lot and she decided to go onto the three-monthly injections of GnRH inhibitor (fewer trips to the doctors), still with the HRT and she has now been on the treatment for about 10 months. It seems to be helping to lower her porphyrin levels and she has been able to put on some weight – she was rather under-weight before. She also got through the summer without an attack, even though it is two years since the last one. However, it has not been as helpful as she had hoped, as her general health is still rather shaky.
There seems to be a great deal of variation in the individual reaction to this treatment. Others have commented that it didn’t work at all, and some people need HRT but cannot tolerate it. So it may be useful, but the only way to find out is to try it and see.
Hormone suppression – update: In May 2006, we published an article on Lynn’s experience of hormone suppression to stop acute attacks. This induces an artificial menopause. Lynn also had to take a ‘safe’ form of HRT to deal with night sweats. Though the treatment had the desired effect of stopping acute porphyria attacks, Lynn found that her health was not good. She became very tired, with the kind of symptoms found in Chronic Fatigue Syndrome. She was unhappy with her job, however, and getting rather depressed. So she decided to leave and attempt a Masters degree.
This was not a success, as she became very ill with a stomach bug about three months after starting. She had to give up the course and took a long time to recover. She has had to live on Incapacity Benefit, which is not easy. Without family support she would probably be deeply in debt by now. She saw a specialist in CFS, who thought that the artificial hormones she was having had probably been the trigger for the fatigue. After some thought, she eventually gave up the hormone treatment. She had been on it for about two years by then, which is the maximum recommended anyway.
Unfortunately, the restart of her natural hormones did not go well. About two months later, she had a severe porphyria attack and had to be treated in hospital. However, that is two years ago and she has not had a severe attack since. She has also started an Open University course (on the subject she wanted to study for a Masters) and is enjoying the challenge. The fatigue is still a persistent problem but the depression has eased. Her experiences suggest that hormone suppression is not a quick answer to repeated acute attacks, though it may be worthwhile in the long term for those with severe problems. It needs to be discussed with both a porphyria specialist and a gynaecologist.
Haem arginate experiences
Stories below are from three patients who regularly receive haem arginate treatments.
Robert
“For the first three years I spent most of my time in hospital, then by chance I heard of this new treatment from a patient on another ward. I told my consultant about this and as a result I was treated with haem arginate, one a day for four days, whilst hospitalised for about a fortnight. The treatment I had whilst having the attack shortened the length of the time I was having to stay in hospital. Even though I had this treatment in hospital I still had regular attacks every month. This went on for two years. So it was suggested by the consultant that I have haem arginate on a weekly basis and have been for the last five years. As a result of this my hospital admissions have been decreased to roughly three a year.”
Kim
“I was given haem arginate throughout my pregnancy as I was under attack during most of it. As soon as I was taken off it my porphyrin levels would shoot straight back up. To make matters worse, they didn’t know what effect it might have on my unborn baby. I had my daughter 4 weeks early and she was quite healthy, without any effect from the haem arginate. The day I had my daughter I had one final dose of haem arginate and then 2 weeks later I had another 2 attacks. I have it once a week now and I feel great. I have a hickman line in now, as prolonged use of haem arginate has caused some problems finding veins for infusions. I know it is expensive but it really does work for me. If I am under attack and I have an infusion, within about 12 hours my pains go away.”
Elizabeth
“My experience with haem arginate has predominantly been great, with many successful outcomes. It seems to be more successful when used early in an attack, the sooner I get the treatment, the less time I seem to spend in pain, and the severity of symptoms is often decreased. I am currently having a one-dose infusion of haem arginate once a week in an attempt to prevent attacks which require hospitalisation. I no longer have more than one attack a month, in fact I have only a few a year and they rarely require hospitalisation. The only real side effect was vein inflammation and phlebitis, which led to many veins in my hands and feet becoming unusable after many years regular use. I am now on my sixth portacath and so I am now running out of suitable veins that can be used on a long-term basis. Despite this, my experience with haem arginate has been very positive, and I don’t think I would be here without it. However, I understand that it has to be used with caution. I also feel that a high carbohydrate diet and good hydration is necessary to work with the drug to maintain its full effect.”